
社内資料:小児統合失調症患者を対象としたプラセボ対照試験【承認時評価資料】
試験概要
タイトル
小児統合失調症患者を対象とした検証的試験
目的
小児の統合失調症患者にロナセン錠8mg/日または16mg/日を6週間投与したときの有効性について、投与6週後のベースラインからのPANSS合計スコア変化量を主要評価項目として、プラセボに対する優越性を検証する。
PANSS: Positive and Negative Syndrome Scale
対象
12歳以上18歳以下の統合失調症患者※1 151例 ※1:DSM-Ⅳ-TRの診断基準により診断
(プラセボ群;47例、8mg/日群;51例、16mg/日群;53例)
選択基準;スクリーニング時および治療期開始前のPANSS合計スコアが60~120
CGI-S評価が3(Mildly)以上の患者 など
DSM-Ⅳ-TR :Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision
PANSS: Positive and Negative Syndrome Scale
CGI-S:Clinical Global Impressions scale-Severity of Illness
試験デザイン
多施設共同、プラセボ対照、無作為化、二重盲検、並行群間比較試験
有効性評価項目
有効性
〔主要評価項目〕
- 投与6週後のベースラインからのPANSS合計スコア変化量
〔副次評価項目〕
- 6週時(LOCF)および各評価時期の各スコア(PANSS尺度別合計スコア、PANSS 5因子モデル別合計スコア、PANSS症状別スコア、CGI-Sスコア)の変化量
- 6週時(LOCF)および各評価時期のPANSSスコアの寛解率※1
- 各評価時期のPANSS合計スコア変化量
- 6週時(LOCF)および各評価時期のCGI-Iの改善率※2
- 治験薬投与開始日から治療期の最終服薬日までの日数
- 6週時(LOCF)および各評価時期でのPANSS responder(PANSS合計スコアがベースラインから20、30、40および50%以上改善した患者)の割合
※1:PANSS評価症状の妄想(P1)、概念の統合障害(P2)、幻覚による行動(P3)、情動の平板化(N1)、受動性/意欲低下による社会的引きこもり(N4)、会話の自発性と流暢さの欠如(N6)、衒奇症と不自然な姿勢(G5)、不自然な思考内容(G9)の「各項目のスコアがすべて3(軽度)以下」になった患者の割合
※2:CGI-Iが1(Very much improved)または2(Much improved)になった患者の割合
PANSS: Positive and Negative Syndrome Scale
LOCF:Last Observation Carried Forward
CGI-S:Clinical Global Impressions scale-Severity of Illness
安全性
- 有害事象および副作用の発現割合
- 錐体外路系有害事象の発現割合
- 6週時(LOCF)および各評価時期のDIEPSS合計スコア変化量(総括重症度を 除く)
- 6週時(LOCF)および各評価時期のDIEPSS症状別スコア変化量
- 6週時(LOCF)および各評価時期の抗パーキンソン薬の併用割合
- 6週時(LOCF)および各評価時期のCGI-SSスコア変化量と悪化率※3
- 臨床検査値、バイタルサインおよび体重、心電図所見および心電図パラメータ変化量
※3:CGI-SSの最新の改善状態が6(Much worse)または7(Very much worse)になった患者の割合
LOCF:Last Observation Carried Forward
DIEPSS:Drug Induced Extra-Pyramidal Symptoms Scale
CGI-SS:Clinical Global Impression of Suicide Severity
解析計画
主要な解析は最大の解析対象集団(FAS)を対象に実施する。
FASは無作為割付けされた患者のうち、以下のすべてを満たす患者の集団とした。
- 治験薬を1回以上投与された患者
- 選択基準である『DSM-Ⅳ-TRの診断基準に基づき統合失調症と診断された患者。なお、M.I.N.I.KIDを補助的に使用する』を満たす患者
- ベースラインおよび投与開始後に測定された少なくとも1つの評価可能なPANSS合計スコアを有する患者
有効性の主要評価項目は、投与6週後のPANSS合計スコアのベースラインからの変化量とし、プラセボ群に対するロナセン錠(8および16mg/日)群の優越性を検証する。
- 主要な解析ではMMRMを用い、共変量として投与群、評価時期、PANSS合計スコアのベースライン、および投与群と評価時期の交互作用を含める。患者内の相関にはUnstructured共分散行列を用い、自由度はKenward-Rogersの近似により算出する。欠測値の補完は行わない。このモデルの反復計算が収束しない場合、共変量の固定効果の標準偏差の推定にサンドウィッチ分散を用い、共分散行列として、heterogeneous Toeplitz、heterogeneous first-order autoregressive、Toeplitz を順に用い、最初に収束したモデルを採用した。
- 検定の多重性は以下の閉検定手順を用いて調整する。Step 1は実薬群合計/2 vs プラセボ群(両側5%)を検定し、有意差が認められた場合、次のStep 2は各実薬群 vs プラセボ群(両側5%)の検定を実施する。
安全性の解析は安全性解析対象集団を対象に実施する。
FAS:Full Analysis Set
M.I.N.I.KID:MINI INTERNATIONAL NEUROPSYCHIATRIC INTERVIEW for Children and Adolescents、PANSS:Positive and Negative Syndrome Scale
MMRM:mixed model for repeated measures
試験デザイン

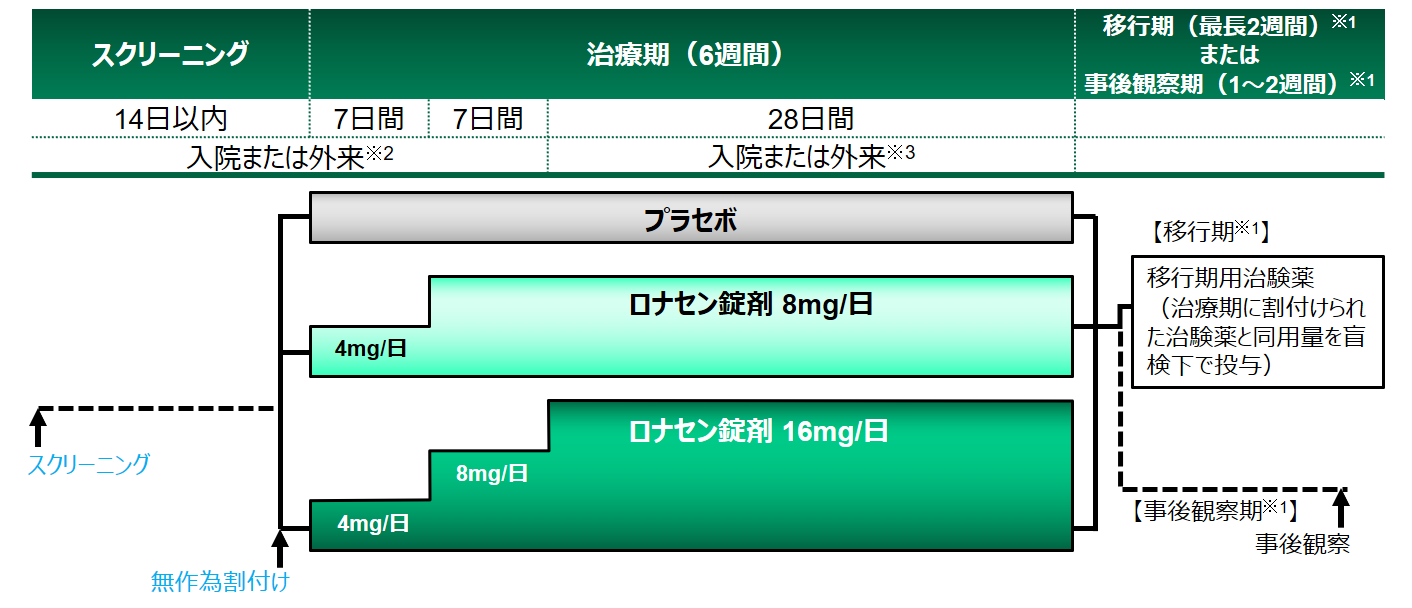
[治療期]
プラセボまたはロナセン錠剤(8mg/日 または 16mg/日)を1日2回に分け、6週間、朝・夕食後に経口投与した。
ロナセン 2mg錠、4mg錠およびそれぞれのプラセボを使用し、各群で錠数が異ならないよう投与した。また、スクリーニング時から事後観察期まで、抗精神病薬・抗パーキンソン薬・向精神薬・睡眠薬・精神療法等の併用は規定に従うこととした。
※1:移行期:長期継続投与試験へ移行した患者、事後観察期:長期継続投与試験へ移行しなかった患者
※2:スクリーニング時のCGI-SS重症度評価スコアが1(Not at all suicidal)で、別途定めた基準を満たした場合、担当医師の判断にて試験期間中は外来観察でよいこととした
※3:スクリーニング時のCGI-SS重症度評価スコアが2以上(Mildly suicidalより悪い状態)の場合、2週後の観察・検査・評価が終了するまでは入院とし、以降は、患者および代諾者から外来への変更の申し出があり、別途定めた基準を満たした場合、退院して外来観察に切り換えてもよいこととした
選択基準
- DSM-Ⅳ-TRの診断基準に基づき統合失調症と診断された患者。なお、M.I.N.I.KIDを補助的に使用する
- スクリーニング時および治療期開始前のPANSS合計スコアが60~120で、CGI-S評価が3(Mildly)以上の患者
- 同意取得(本人および代諾者)時の本人の年齢が12歳以上18歳以下の患者
- 治験の目的、予測される薬効・薬理作用および危険性などについて、患者および代諾者が十分に説明をうけ、理解が得られ、患者の治験参加について本人および代諾者から 自由意思による同意を文書で得られた患者(本人からはアセント文書にて同意取得)
主な除外基準
- ロナセンの投与を受けたことのある患者
- ロナセン経口剤の添付文書で禁忌に該当する状況の患者
- 悪性症候群、遅発性ジスキネジア、麻痺性イレウス、横紋筋融解症、無顆粒球症、肺塞栓症、深部静脈血栓症の合併または既往歴がある患者
- パーキンソン病の患者
- 強い自殺念慮を有する患者、自殺手段としての自傷行為、自殺企図の既往のある患者
- 糖尿病の患者
- 重篤な心血管系疾患、肝疾患、腎疾患、脳器質性疾患、血液疾患、内分泌疾患、痙攣性疾患などの合併症を有する患者
- クロザピンの服用歴がある患者またはスクリーニング時の前1年(365日)以内に2種類以上の抗精神病薬を十分量服用しても精神症状が改善しなかったと治験責任医師または治験分担医師が判断した患者
- スクリーニング時の前3ヵ月(90日)以内に抗精神病薬のデポ剤(持続性製剤)の投与を受けた患者
- スクリーニング時の前6ヵ月(180日)以内に電気痙攣療法を受けた患者
- 妊婦、妊娠している可能性のある患者 など
症例の内訳


16mg/日群で「ベースラインのPANSS合計スコアがない患者」が1例おり、FASから除外した。よって、FASの患者数は、 プラセボ群、8/日群および16mg/日群の各群で、それぞれ47例、51例、52例となった。