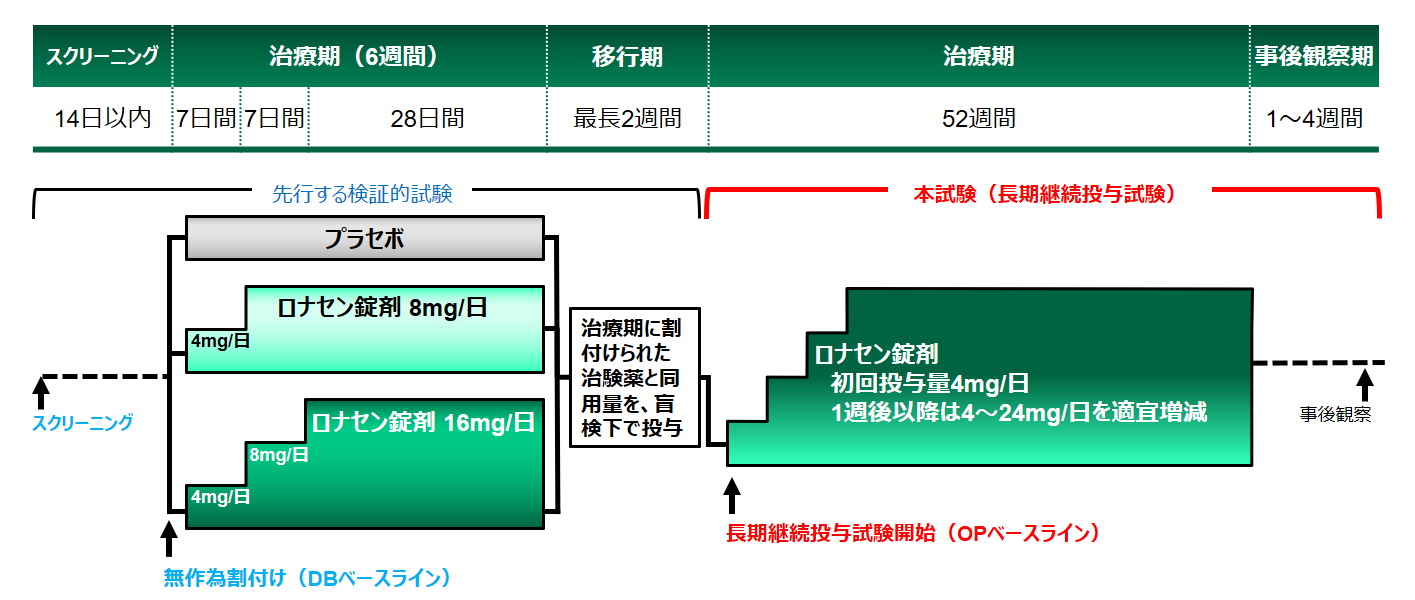
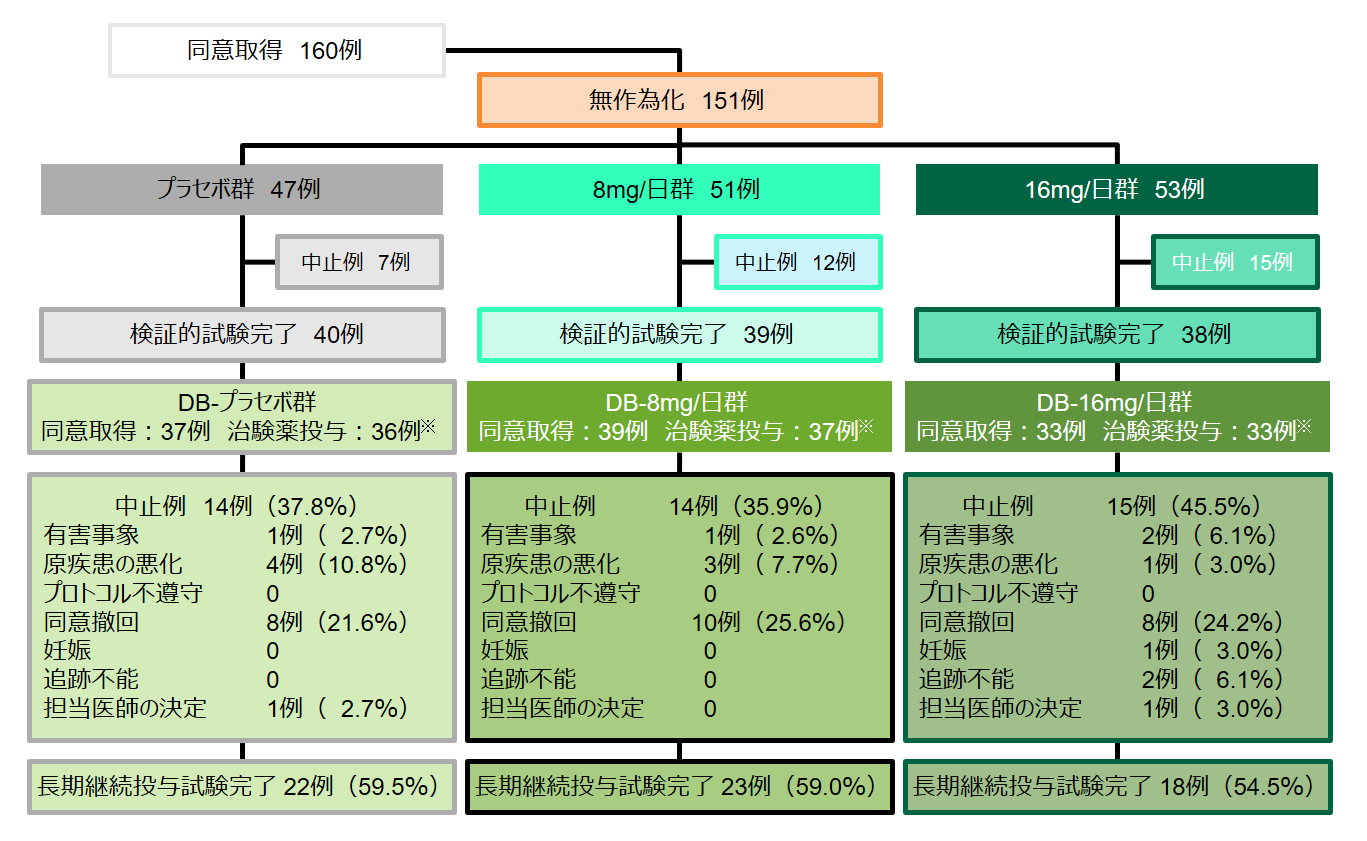
有効性
〔主要評価項目〕
- 52週時(LOCF)の各ベースラインからのPANSS合計スコア変化量
〔副次評価項目〕
- 52週時(LOCF)および各評価時期の各スコア(PANSS尺度別合計スコア、PANSS 5因子モデル別合計スコア、PANSS症状別スコア、CGI-Sスコア)の変化量 各評価時期のPANSS合計スコア変化量
- 52週時(LOCF)および各評価時期のPANSSスコアの寛解率※1
- 52週時(LOCF)および各評価時期のCGI-Iの改善率※2
- 本試験の治験薬投与開始日から最終服薬日までの日数
- 52週時(LOCF)および各評価時期でのPANSS responder(PANSS合計スコアがベースラインから20、30、40および50%以上改善した患者)の割合
※1:PANSS評価症状の妄想(P1)、概念の統合障害(P2)、幻覚による行動(P3)、情動の平板化(N1)、受動性/意欲低下による社会的引きこもり(N4)、会話の自発性と流暢さの欠如(N6)、衒奇症と不自然な姿勢(G5)、不自然な思考内容(G9)の「各項目のスコアがすべて3(軽度)以下」になった患者の割合
※2:検証的試験の治療開始時からの比較でCGI-Iが1(Very much improved)または2(Much improved)になった患者の割合
LOCF:Last Observation Carried Forward
PANSS: Positive and Negative Syndrome Scale
CGI-S:Clinical Global Impressions scale-Severity of Illness
CGI-I:Clinical Global Impressions scale-Global Improvement
安全性
- 有害事象および副作用の発現割合
- 錐体外路系有害事象の発現割合
- 52週時(LOCF)および各評価時期のDIEPSS合計スコア変化量(総括重症度を�除く)
- 52週時(LOCF)および各評価時期のDIEPSS症状別スコア変化量
- 52週時(LOCF)および各評価時期の抗パーキンソン薬の併用割合
- 52週時(LOCF)および各評価時期のCGI-SSスコア変化量と悪化率※3
- 臨床検査値、バイタルサインおよび体重、心電図所見および心電図パラメータ変化量
※3:CGI-SSの最新の改善状態が6(Much worse)または7(Very much worse)になった患者の割合
DIEPSS:Drug Induced Extra-Pyramidal Symptoms Scale
CGI-SS:Clinical Global Impression of Suicide Severity