
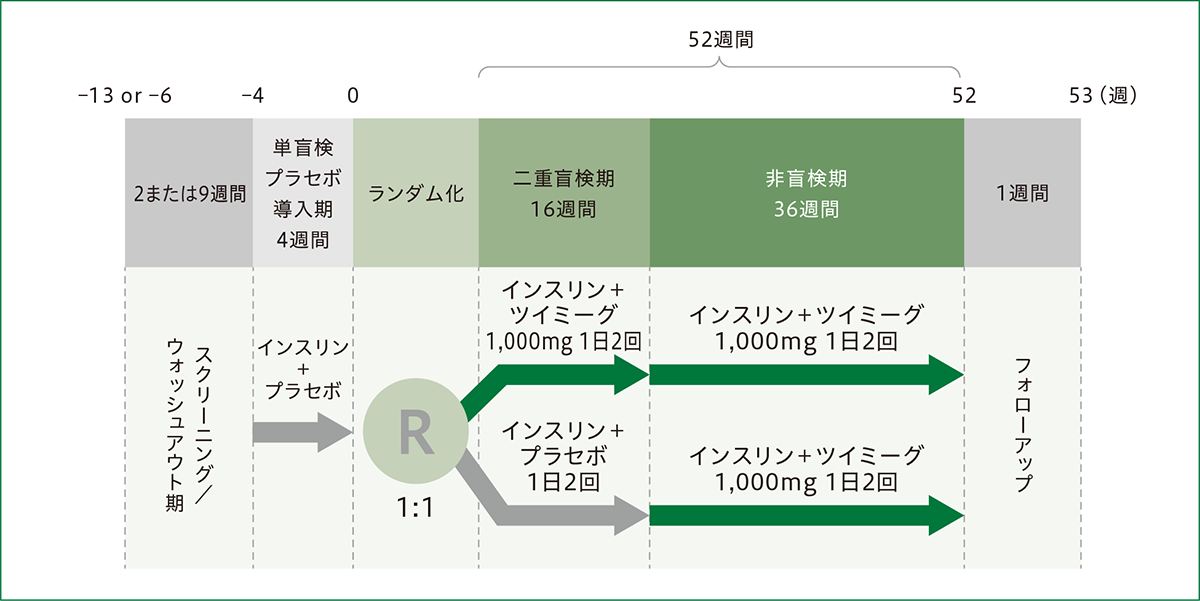
| 【試験デザイン】 | 二重盲検期(16週間);第3相、多施設共同、無作為化、二重盲検、プラセボ対照、並行群間比較試験 非盲検期(36週間);非盲検、非対照試験 |
| 【目的】 | 二重盲検期;〔主要目的〕インスリン治療※1による血糖コントロールが不十分※2な日本人2型糖尿病患者を対象として、ツイミーグ1,000mgを1日2回(2,000mg/日)経口投与とインスリンを併用投与したとき、投与16週間後のHbA1cの改善をインスリン単独投与と比較して評価する。 非盲検期;〔副次目的〕ツイミーグとインスリンの併用投与が投与52週間後のHbA1cを改善するか評価する。 |
| 【対象】 | インスリン治療※1で、血糖コントロールが不十分(7.5%≦HbA1c<11.0%※2)な日本人2型糖尿病患者215例(20歳以上、eGFR※3≧60mL/min/1.73m2)[FAS集団:214例、安全性解析対象集団:215例、非盲検安全性解析対象集団:209例] |
| 【方法】 | スクリーニング期(インスリン単独療法を受けていた患者では2週間、インスリンと1種類の経口血糖降下薬による併用療法を受けていた患者では1週間のスクリーニングおよび8週間のウォッシュアウト)および4週間の単盲検プラセボ導入期の後、患者はインスリン併用下において、1:1の比でプラセボまたはツイミーグ1回1,000mgのいずれかに無作為化され、16週間の二重盲検期を開始した。その後、36週間の非盲検期を継続した。なお、無作為化のとき、前治療状況(インスリン単独療法、インスリン+経口血糖降下薬1剤の併用療法)および無作為化前来院時のHbA1c(8.0%未満、8.0%以上)を層別因子とした。 二重盲検期(16週間);インスリン併用下で、プラセボまたはツイミーグ1回1,000mgを朝、夕の1日2回(2,000mg/日)16週間経口投与 非盲検期(36週間);インスリン併用下で、ツイミーグ1回1,000mgを朝、夕の1日2回(2,000mg/日)36週間経口投与 なお、ツイミーグは、朝および夕の食事中または食事直後の服用を推奨とした。また、インスリンの1日投与量は、二重盲検期では一定(ベースラインの1日投与量の±10%未満、ただし安全上の理由により用量調節が必要な場合の減量は可能)に維持し、非盲検期では調節可能とした。 |
| 【主要評価項目】 | 16週時のHbA1cのベースラインからの変化量※4(検証的解析項目) |
| 【重要な副次評価項目】 | 16週時のHbA1c改善目標達成割合※5 |
| 【その他の副次評価項目】 | 二重盲検期(16週間);16週時のベースラインからの変化量※4(空腹時血糖、総コレステロール、LDLコレステロール、HDLコレステロール、トリグリセリド、hsCRP)、16週時のインスリン平均1日投与量のベースラインからの変化量※4、16週時のインスリン平均1日投与量のベースラインからの減少率が10%超の患者割合※4、レスキュー治療患者割合※6 非盲検期(36週間)/治療期全体(52週間);投与終了時のHbA1c、空腹時血糖、総コレステロール、LDLコレステロール、HDLコレステロール、トリグリセリド、hsCRPのベースライン(ツイミーグ初回投与前)からの変化量※4、投与終了時点でのHbA1c改善目標達成割合※5、投与終了時のインスリン平均1日投与量のベースライン(同)からの変化量※4、投与終了時のインスリン平均1日投与量のベースライン(同)からの減少率が10%超の患者割合※4、レスキュー治療患者割合※6 |
| 【安全性評価項目】 | 有害事象、副作用 等 |
| 【解析計画】 | 二重盲検期(16週間);有効性評価項目はいずれもFAS集団を対象に解析した。 有効性の主要評価項目は、主解析として、投与群、評価時点、層別時の前治療状況、投与群と評価時点の交互作用を固定効果、ベースライン時のHbA1cを連続共変量とし、無構造の分散共分散構造を仮定したMMRMを用いて、ツイミーグ+インスリン群とプラセボ+インスリン群の最小二乗平均値の差と95%信頼区間、並びにp値を算出した。加えて、各評価時点のHbA1cおよびベースラインからの変化量の要約統計量も算出した。投与16週までに治験薬を中止した場合、主解析には投与中止後7日までのデータを用いた。HbA1c(%)のベースラインからの変化量の最小二乗平均を95%信頼区間と共に投与群および評価時点(4週おき)別にグラフで示し、経時的な傾向を検討した。 有効性の重要な副次評価項目の解析では、投与群、層別時の前治療状況を固定効果、ベースライン時のHbA1cを連続共変量としたロジスティック回帰モデルを用いて解析し、ツイミーグ+インスリン群とプラセボ+インスリン群のオッズ比とその95%信頼区間、並びにp値を算出した。治験薬投与を中止した場合を含め、投与16週時のHbA1cを有しない患者は、目標未達成として集計した。なお、主要評価項目と副次評価項目間での検定の多重性は、主要評価項目で有意であった場合のみ副次評価項目の検定を実施する固定順序法にて調整した。 その他の副次評価項目(空腹時血糖等)では、投与群、評価時点、層別時の前治療状況およびHbA1c、投与群と評価時点の交互作用を固定効果、各ベースライン値を共変量としたMMRMを用いて、主要評価項目と同様の解析を実施した。 二重盲検期における主要評価項目について、腎機能( CKD1:eGFR≧90mL/min/1.73m2、CKD2:60≦eGFR<90mL/min/1.73m2)・年齢(65歳未満、65歳以上)等の部分集団解析(MMRM)を実施した。 非盲検期(36週間)/治療期全体(52週間);有効性評価項目は非盲検安全性解析対象集団を対象に解析し、ベースラインは、二重盲検期でツイミーグ+インスリン群であった患者(ツイミーグ/ツイミーグ群)では、二重盲検期の初回投与前のデータ、二重盲検期でプラセボ+インスリン群であった患者(プラセボ/ツイミーグ群)では、非盲検期の初回投与前のデータとした。治験薬投与を中止した場合は、投与中止後7日までのデータを用いた。長期の有効性解析(HbA1c等、連続変数)では、各ベースライン値を連続共変量、評価時点、層別時の前治療状況を固定効果とし、無構造の分散共分散構造を仮定したMMRMを用いて、最小二乗平均値と95%信頼区間を算出した。さらに、各評価時点の測定値およびベースラインからの変化量の要約統計量を投与群ごとに算出した。また、カテゴリカル解析として、要約統計量を算出した。 |
※1:インスリン(基礎インスリンまたは混合インスリンを1日2回投与まで、1日投与量が8IU/日以上40IU/日以下)単独療法またはインスリン(同)と経口血糖降下薬1剤の併用療法
※2:インスリン単独療法の場合;スクリーニング時および無作為化前時7.5%≦HbA1c<11.0%
インスリンと経口血糖降下薬併用療法の場合;スクリーニング時7.5%≦HbA1c≦10.0%、無作為化前時7.5%≦HbA1c<11.0%
※3:無作為化前時日本人用MDRD(Modified Diet in Renal Disease)式による推算糸球体濾過量
※4:治験薬の投与を中止した場合は投与中止7日後までのデータを使用
※5:HbA1cが7.0%未満を達成した患者の割合、HbA1cのベースラインからの相対減少率が7%以上であった患者の割合。ただし、治験薬の投与を中止した場合は投与中止7日後までのデータを使用
※6:二重盲検あるいは非盲検期中に、高血糖またはHbA1c高値となったためにレスキュー治療を要し、治験薬の投与を中止した患者の割合
| 10. 相互作用(抜粋) 10.2 併用注意(併用に注意すること) 薬剤名等:糖尿病用薬(インスリン製剤、スルホニルウレア剤、速効型インスリン分泌促進薬、α-グルコシダーゼ阻害剤、チアゾリジン系薬剤、DPP-4阻害剤、GLP-1受容体作動薬、SGLT2阻害剤 等)[11.1.1参照]、ビグアナイド系薬剤[8.5、11.1.1参照] |










